
 返回首页
返回首页实验室在金属催化剂表面键合分子助剂及其电子效应取得新进展
 时间 :2022年09月02日 来源于:煤转化国家重点实验室
时间 :2022年09月02日 来源于:煤转化国家重点实验室

时间 :2022年09月02日 来源于:煤转化国家重点实验室
实验室张斌副研究员、覃勇研究团队与刘星辰副研究员合作,实现了通过一种Pt纳米颗粒表面自限制反应构筑稳定CpCo-Pt化学键新策略。简单改变CpCo-在Pt表面的覆盖度,即可梯度定量调控Pt纳米颗粒表面电子密度。研究明确了Pt颗粒表面电子密度的增加源于Co d轨道和Cp的π轨道。这种远程的电子效应减弱了烯烃的吸附并增加了Pt上C=C加氢的活化能,进而提高炔烃半加氢制烯烃的选择性。该成果近日以Direct Bonding of CpCo- Fragments on Pt Nanoparticles and their Electronic Effect for Alkyne Semihydrogenation为题发表在《ACS Catalysis》上。

研究利用ALD在SiO2纳米线上沉积Pt纳米颗粒,再向其表面脉冲二茂钴(CoCp2),通过表面自限制方法,CpCo-分子与Pt表面直接形成化学键(图1a)。HRTEM、EDX-Mapping、原位红外和XAFS等多种表征结果证明了CpCo-分子在Pt颗粒表面高度分散,所制备的催化剂保留了CoCp2的分子结构和配位环境,并且形成了CpCo-Pt化学键(图1b-e)。
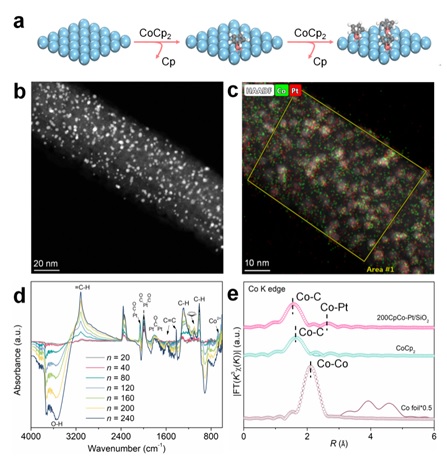
图1 催化剂制备过程示意图、表面形貌和结构
进一步通过红外CO化学吸附、XPS和DFT计算确定了CpCo-分子修饰Pt纳米颗粒后,Pt表面电子密度增加,CpCo-分子通过形成CpCo-Pt化学键进行电子传递,CpCo-覆盖度与Pt表面电子密度的增加存在线性关系(图2)。
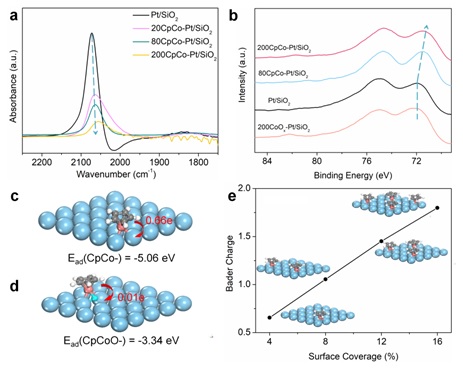
图2 CpCo-修饰对于Pt的电子效应和DFT计算
通过投影态密度分析(PDOS)来了解CpCo-分子和Pt(111)之间巨大电荷转移背后的确切轨道相互作用。进一步分析表明Cp π轨道和一小部分Co d轨道的电子主要转移到Pt的s、pz和d轨道,部分到Co的s和p轨道(图3)。
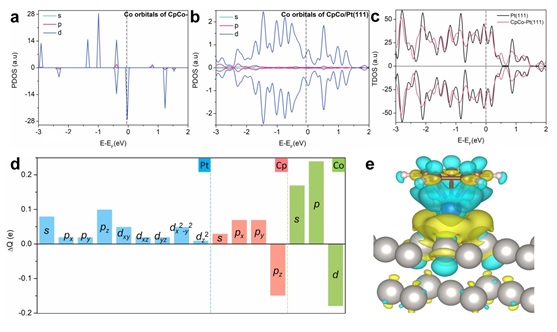
图3 分子助剂电子效应及分子-金属电子传递轨道分析
将制备的催化剂用于苯乙炔半加氢反应中,发现CpCo-修饰后的催化剂可以显著提高苯乙炔制苯乙烯的选择性。拉曼、红外和动力学测试结果证明CpCo-修饰会促进苯乙烯在Pt表面的脱附,并增加了C=C加氢的活化能(图4)。
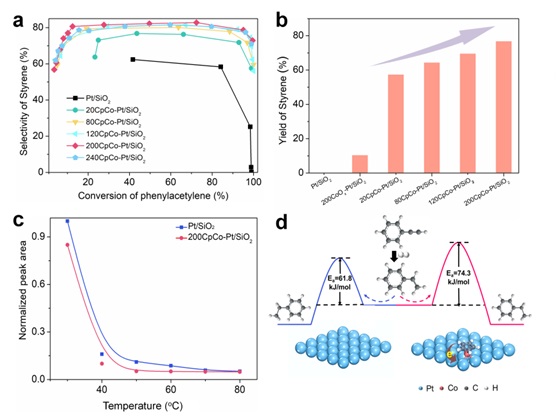
图4 炔烃半加氢反应性能和选择性提升机制
这项工作通过设计具有清晰结构、位置和组成的助剂,为设计具有可量化电子密度的高效催化剂以进行选择性加氢提供了可行的方案。该工作得到了国家自然科学基金、国家杰出青年科学基金、国家重点研发计划、中科院青年促进会优秀会员、榆林学院与大连清洁能源国家实验室联合基金、山西省重大科技专项、山西省自然科学基金、煤转化国家重点实验室青年人才培养计划、北京光源的资助与支持。
原文链接: https://pubs.acs.org/doi/full/10.1021/acscatal.2c03024
(903组)
 委员会信箱
委员会信箱 官方微信
官方微信 官方微博
官方微博 地址:中国·山西省·太原市迎泽区桃园南路27号
地址:中国·山西省·太原市迎泽区桃园南路27号 邮编:030001
邮编:030001